开展一些AI-assist Drug Design 的方向探索,需要进行一些论文阅读和调研,形成调研报告。
参考:
- [2402.08703] A Survey of Generative AI for de novo Drug Design: New Frontiers in Molecule and Protein Generation
- Hao Zhou(周浩)
目标:
- 主要解决什么问题
- 挑战是什么
- 我们提出的核心方法,与同类问题比较的优势在哪
- 数据集是什么,是否公开
- 评测方式是什么,有无数据集
思维导图:
A Survey of Generative AI for de novo Drug Design: New Frontiers in Molecule and Protein Generation
摘要
将从头药物设计(de novo drug design)归纳为两大主题:小分子生成和蛋白质生成。在每个主题下,我们识别出多种子任务和应用,重点介绍重要的数据集、基准测试、模型架构,并比较顶尖模型的性能。
引言
- 并非是虚拟筛选/定向进化。而是从头开始的自然界中并未存在的新的生物实体的生成。
- 文章中分成两个部分来讲解。小分子和蛋白质。
- 文章将介绍
- 生成式模型的种类:Diffusion/VAE/Flow-Based/GAN
- 将文章分成小分子和蛋白质两个领域,分别介绍
- 一般背景/任务定义
- 用于训练和测试的常见数据集
- 常用的评估指标
- 对过去和当前的机器学习方法的概述
- 对SOTA方法的性能的对比分析
- 总结
相关研究
其他的方法都太专业,而这篇文章对小分子和蛋白质生成进行宏观层面的分析,有助于那些想要对化学创新领域中新兴的生成式AI模型有一个高屋建瓴的了解的人。
前言:生成式AI模型
介绍了VAE、GAN、Flow-Based Models和Diffusion,还介绍了一些其他模型,比如GNN、EGNN等。
应用
小分子
任务背景
分子生成聚焦于为药物设计创造新的分子化合物。这些生成的分子旨在具有 (1)有效性、(2)稳定性和(3)独特性,总体目标是具有药物适用性。“药物适用性” 是一个宽泛的术语,用于描述分子对各种生物靶标的结合亲和力。
虽然前三个任务可能看起来微不足道,但仅仅生成有效和稳定的分子就存在各种挑战。因此,无靶向分子生成领域专注于生成有效的分子集合,而不考虑任何生物靶标。靶向分子生成(或配体生成)侧重于针对特定蛋白质结构生成分子,因此更关注药物成分。最后,3D 构象生成涉及在给定 2D 连接的情况下生成各种 3D 构象。
无靶向分子设计
必须满足前两个特点,也就是有效性和稳定性。需要满足很多的复杂的条件,所以其实还是挺难的。深度学习可以帮助人们更有效率地生成有更高可能性满足有效性的分子。
任务:无输入,输出为生成一组新的、有效的、稳定的分子。
数据集:QM9和GEOM-Drug
指标:
- 分子生成任务指标:原子稳定性、分子稳定性、有效性、独特性、(新颖性)、药物相似性度量估计值QED。
- 模型的评估方式:通过在QM9数据集上的一部分训练属性分类网络,然后对模型生成的分子进行评估,计算目标和评估属性值之间的平均绝对误差。
- 分子在特定化学性质方面的指标:极化率$α$、最高占据分子轨道能量 $\varepsilon_{HOMO}$、最低未占据分子轨道能量 $\varepsilon_{LUMO}$、$\varepsilon_{HOMO}$ 和 $\varepsilon_{LUMO}$ 的差值$\Delta_\varepsilon$、偶极矩 $\mu$、298.15K 下的摩尔热容 $C_v$。
模型:
过去几年间,分子生成任务的方法从一维的SMILES转变成二维的连接图,然后是三维的几何结构,最后到融合二维和三维信息的方法。
- 1D SMILES字符串模型
- 早期方法:如CVAE、GVAE直接处理,但是SMILES因为是一维的,存在问题:两个化学结构相似的分子图可能会得到非常不同的 SMILES 字符串,这使得模型更难学习到这些相似性和模式。
- 2D 图生成模型
- JTVAE:首个直接生成 2D 分子图的模型,通过树状骨架迭代扩展并验证结构有效性。
- 3D 结构模型
- 早期方法:Flow-Based方法ENF和自回归方法G-SchNet。
- EDM:基于Diffusion的 3D 点云模型,利用 E (3) 等变性提升性能,避免原子排序依赖。
- GCDM:结合几何深度学习与Diffusion,引入注意力机制优化消息传递。
- 联合2D和3D
- JODO:联合2D和3D的扩散模型,使用几何图形表示来捕获 3D 空间信息和连接信息,对这种联合表示应用分数随机微分方程,同时提出扩散图变换器来参数化数据预测模型,避免在每个独立通道独立添加噪声后相关性的丢失。
- MiDi:应用了DDPM,提出了「松弛」的图神经网络(EGNN)。
这里给出了三个表格。
- 表格1:生成模型在QM9数据集上的条件无关的分子设计任务上的性能表现。Diffusion方法比之前的方法好很多,但是在GEOM-Drugs上可能表现不佳。
- 表格2:EDM、MDM、MiDi 等模型在GEOM-Drugs数据集上的条件无关的分子设计任务上性能表现。MiDi能生成更稳定的复杂分子,但是有效性较低。
- 表格3:生成模型在条件分子生成任务上的性能表现。MDM、GCDM生成表现不错,MDM前四项较好,GCDM后两项较好。
靶向分子设计
有两种,一种是基于配体的药物设计(LBDD),另一种是基于结构的药物设计(SBDD)。LBDD利用目标蛋白质的氨基酸序列,借助已知的配体特征来构建;SBDD利用目标蛋白质的三维结构来设计。
任务:给定氨基酸序列/蛋白质的三维结构,生成对应的有高结合亲和力以及潜在相互作用的分子。
数据集:CrossDocked2020、ZINC20和Binding MOAD。
指标:Vina Score、Vina Energy、高亲和力百分比High Affinity Percentage、合成可及性分数SAscore和多样性Diversity。
模型:
- LBDD
- 结合了Transformer结构,例如DrugGPT,训练的时候输入为SMILES和蛋白质氨基酸序列,从而训练输出可行的SMILES配体。
- SBDD
- LiGAN:三维目标感知分子输出的概念,将分子适配到网格格式,以便利用卷积神经网络(CNN)进行学习,并在变分自编码器(VAE)框架下训练模型
- TargetDiff模型:基于EGNN进行Diffusion,在结构上和EDM相似,目标是学习条件分布。特别地,研究人员通过原子嵌入的熵来将原子类型的灵活性降低,从而提高结合亲和力。
- DiffSBDD:DiffSBDD-cond是一种DDPM,而在基准测试中,DiffSBDD-inpaint则进行了图像增强,使用掩蔽和替换等方法对配体-蛋白质的部分区域进行处理。
这里给出了一个表格。展示了不同的模型的结果。
蛋白质
任务背景
蛋白质可以通过其3D结构或者氨基酸序列来表示,氨基酸序列类似人类的语言,可以应用于自然语言模型。可以定义几个子任务:1)表示学习。2)结构预测。3)序列生成。4)主干设计。此外还讨论了抗体生成和肽生成。
蛋白质表示学习
使用氨基酸序列/原子坐标学习一个嵌入从而为其他生成模型创建更丰富的数据空间以供训练。类似于自然语言处理中的word2vec。
结构预测
从氨基酸序列来预测结构是极具挑战性和重要的工作。
任务:从氨基酸序列来预测蛋白质结构。
数据集:主要来自蛋白质结构预测的关键评估(CASP)。有PDB、CASP14和CAMEO。
指标:均方根误差RMSD、全局距离测试总得分GDT-TS、模板建模得分TM-score和局部距离差异测试LDDT。
模型:
- AlphaFold2:里程碑模型。采用端到端的方式集成了多层Transformer,融合多序列比对和成对表示的信息。基于氨基酸之间的成对距离探索折叠空间、氨基酸的潜在取向和整体结构。
- trRosetta:transform-restrained Rosetta。输入MSA之后,预测残基对之间的距离和取向,然后利用Rosetta协议构建3D结构。
- RoseTTAFlod:在CASP14上表现效果比肩AlphaFold2,特别是生成速度很快,仅需10分钟,相较AlphaFold2快了100倍。
- ESMFold:利用ESM-2的输出嵌入到自注意力「折叠块」中,并通过以哦个具有SE(3)的transformer架构的结构模块生成最中国的结构预测。
- EigenFold:应用Diffusion生成蛋白质结构的模型。它将蛋白质表示为一个谐振子系统,在正向过程中可以将结构投影到该系统的本征模式上,在反向过程中先采样粗糙的全局结构再细化局部细节。作为一种基于分数的模型,EigenFold 计算强度不高,但在准确性和范围方面仍不如其他模型。
抗体结构预测:
这里的MSA结构不能作为抗体的输入。因此通用的模型如AlphaFold2效率非常低。
- IgFlod:使用来自AniBERTy的序列嵌入和不变点注意力机制来预测。
- tFlodAb:减少了对Rosetta能量函数等外部工具的依赖。
序列生成
序列生成,也被称为反向折叠或固定骨架设计,是结构预测的逆任务。生成能折叠成目标结构的氨基酸序列,对于设计具有期望结构和功能特性的蛋白质至关重要。由于有效序列的空间巨大,且蛋白质折叠过程复杂难以预测,因此需要多种深度学习方法来解决这些挑战
任务:给定固定的蛋白质骨架结构,生成能折叠成该结构的相应氨基酸序列。
数据集:模型主要使用 CATH 进行训练,部分会利用 UniRef 和 UniParc 进行数据增强,评估时常用 CATH 和 TS500。此外,Yu 等人创建了一组 14 个已知的从头蛋白质结构,用于避免数据污染。
指标:
- AAR(氨基酸恢复率):生成序列与天然序列中匹配氨基酸的比例。
- 多样性(Diversity):通过 Clustalw2 测量生成序列对之间的平均差异。
- RMSD(均方根偏差):将生成序列折叠成结构后,与天然骨架结构进行比较的结构差异指标。
- 非极性损失(Nonpolar Loss):衡量折叠结构中极性氨基酸类型合理性的指标,表面非极性氨基酸含量越高,损失越大。
- PPL(困惑度):交叉熵损失的指数化,代表天然序列出现在预测序列分布中的逆可能性。
模型:
- 初步的一类模型在不考虑固定骨架目标的情况下生成蛋白质序列。但这些模型无法考虑关键的结构信息。
- ProteinVAE 利用 ProtBERT 将原始输入序列转化为潜表示;
- ProT-VAE 使用不同的预训练语言模型 ProtT5NV;
- ProteinGAN 则采用 GAN 架构。
- 主要的模型接收固定骨架目标作为输入来生成氨基酸序列。
- ProteinSolver 将生成骨架结构与解决数独问题联系起来,使用 GNN 架构;
- PiFold 引入更全面的特征表示;
- Anand 等人设计 3D CNN 直接学习条件分布;
- ABACUS-R 结合预训练的 transformer 来推断残基的氨基酸类型;
- ProRefiner 通过引入熵分数改进预测。
- GPD 使用 Graphormer 架构,
- GVP-GNN 采用新颖的几何表示,
- ESM-IF1 扩展表示并在扩展数据集上训练,
- ProteinMPNN 实现了顺序无关的自回归方法。
- 在这些模型中,ProteinMPNN 在序列恢复、RMSD 和非极性损失方面表现最佳,GPD 则是最省时的方法。
主干设计
生成全新的蛋白质可以直接扩充蛋白质库,实现高度复杂和多样的功能,是从头设计的核心。蛋白质设计在结构和序列上存在差异,有的模型生成 1D 氨基酸序列,有的直接生成 3D 结构,还有的同时设计两者。
任务:从无输入或基于现有背景设计蛋白质骨干结构,即生成每个氨基酸的骨干原子(氮、$\alpha$ - 碳、羰基和氧原子)坐标,外部工具可用于侧链填充。包含上下文无关生成(生成多样的蛋白质结构)和上下文给定生成(根据天然蛋白质的基序填充缺失残基)两个子任务。
数据集:常用的数据集有 PDB、AlphaFoldDB、SCOP(及其扩展 SCOPe)和 CATH。
指标:
- scTM(自洽 TM 分数):通过将提议的结构输入序列预测模型(通常是 ProteinMPNN)生成相应氨基酸序列,再将其输入结构预测模型(通常是 AlphaFold2)生成样本结构,计算生成结构与样本结构之间的 TM 分数。分数大于 0.5 的结构通常被认为是可设计的。
- scRMSD(自洽 RMSD):与 scTM 类似,但使用 RMSD 进行评估,分数小于 2 通常作为截止值。
- AAR(氨基酸恢复率):比较生成的氨基酸序列与真实序列的相似程度。
- RMSD(均方根偏差):衡量生成的残基坐标与真实值之间的距离。
模型:
- 较短蛋白质
- ProtDiff 使用 3D 笛卡尔坐标表示每个残基和粒子滤波扩散方法,但 3D 笛卡尔点云不能反映蛋白质折叠过程;
- FoldingDiff 则使用角度表示,更接近蛋白质折叠过程中的旋转能量优化,通过 DDPM 和 BERT 架构从随机未折叠状态去噪到折叠结构;
- LatentDiff 先使用带 GNN 的等变蛋白质自动编码器将蛋白质嵌入潜在空间,再用等变扩散模型学习潜在分布,在潜在空间采样比在原始蛋白质空间快十倍。
- 长蛋白结构:基于框架的构建方法
- Genie 使用由平移和旋转元素确定的框架云进行离散时间扩散来生成骨干结构;
- FrameDiff 基于框架流形参数化骨干结构,使用基于分数的生成模型;
- RFDiffusion 结合 RoseTTAFold 的强大结构预测方法和扩散模型,通过微调 RoseTTAFold 权重并输入掩码输入序列和随机噪声坐标来迭代生成骨干结构,还进行自我条件约束,性能优异;
- GPDL 使用 ESMFold 代替 RoseTTAFold 作为基础结构预测模型,并结合 ESM2 语言模型提取进化信息,生成骨干结构速度比 RFDiffusion 快 10 - 20 倍。
- 同时设计蛋白质序列和结构
- GeoPro 使用 EGNN 编码和预测 3D 蛋白质结构,并设计单独的解码器解码蛋白质序列;
- Protpardelle 在反向扩散过程中对可能的侧链状态进行 “叠加” 并在每次迭代更新时进行塌缩;
- ProtSeed 使用三角函数感知编码器计算约束和相互作用,并通过等变解码器更新序列和结构;
- Anand 等人使用 IPA 在框架空间中进行扩散,高效生成蛋白质序列和结构 。
抗体CDR-H3生成:
特别地,抗体生成聚焦于一个被称为 CDR-H3 区域的生成。最开始使用的是LSTM方法,后来转变为RefineGNN方法。此外,一些模型超越了CDR-H3生成任务,而是一次性处理抗体生成的多个环节。dyMEAN是一种端到端的方法将结构预测、对接和CDR-H3生成整合到一个模型中。
多肽设计
虽然已经有在蛋白质生成方面的重要、强大的模型,但是由于多肽结构的复杂和依赖于上下文已经下游应用的多样性,因此有必要为多肽的需求来定制模型。
多肽生成:从头生成新型多肽
- MMCD:基于Diffusion的治疗性多肽生成模型,它联合设计多肽序列和结构(骨干坐标),采用Transformer编码器处理序列,EGNN 处理结构,并运用对比学习策略对齐序列和结构嵌入,区分治疗性和非治疗性多肽嵌入。
多肽-蛋白质互相作用:预测提议的多肽 - 蛋白质对的物理结合位点
- PepGB:基于GNN的模型。它利用图注意力神经网络学习多肽和蛋白质之间的相互作用。
多肽表示学习:将原始多肽序列转换为能捕获有价值信息的潜在表示
- PepHarmony:使用序列编码器(ESM)和结构编码器(GearNet),多视图对比学习模型,集成序列和结构信息以增强多肽表示学习。
多肽测序:解决质谱分析中从含噪数据提取氨基酸序列的挑战
- AdaNovo:从头多肽测序模型,由质谱编码器和两个受Transformer架构启发的多肽解码器组成。它利用条件互信息和自适应训练策略,在多种物种的多肽水平和氨基酸水平精度上显著优于之前的模型。
最近趋势
生成式AI正在深刻地改变药物设计。
- 生成式AI领域:GNN和基于图的方法的出现,推动了从基于序列的方法向基于结构的方法的转变,最终促使在生成任务中实现了序列和结构的整合。
- 分子生成领域:基于图的Diffusion模型作为主导。利用E(3)等变形来实现最先进的性能。
- GeoLDM、MiDi——无靶点分子设计
- TargetDiff、Pocket2Mol、DiffSBDD——有靶点分子设计
- Torsional Diffusion——分子构象生成
- 此外,有靶点分子设计中也出现了从基于序列的方法到基于结构的方法的出现。
- 蛋白质生成领域:也出现了从序列到结构的转变。
- GearNET:基于结构的表示学习模型
- ESM-1B、UniRep:3D结构的重要性
- AlphaFold2:结构预测的最先进模型
- 一些Diffusion方法也致力于蛋白质骨架构建。
挑战
分子生成领域:
- 复杂性
- 适用性
- 可解释性
蛋白质生成领域:
- 基准测试
- 性能
结论
介绍了生成式AI在从头开始的药物设计上的全貌,特别关注分子和蛋白质生成。
Regularized Molecular Conformation Fields | NeurIPS 2022
Regularized Molecular Conformation Fields | OpenReview
属于上面综述文章里里面的分子生成里面的3D构象生成的部分。
解决的问题
给定2D分子图,预测有机分子能量最有利的3D构象。
挑战
- 分子在三维欧氏空间的 SE (3) 变换下具有不变性,同一分子的构象在刚性运动下有无限可能,增加了建模难度
- 分子在环境条件下存在多种动力学,导致高维且复杂的势能面,使得机器学习模型难以识别局部最小值来生成能量有利的构象
- 现有方法使用的不变特征可能冗余、相互依赖,导致数值不稳定和不合理的构象预测,且专门的等变层可能降低神经网络的表达能力,部分模型处理环状图存在困难
核心方法和优势
核心方法
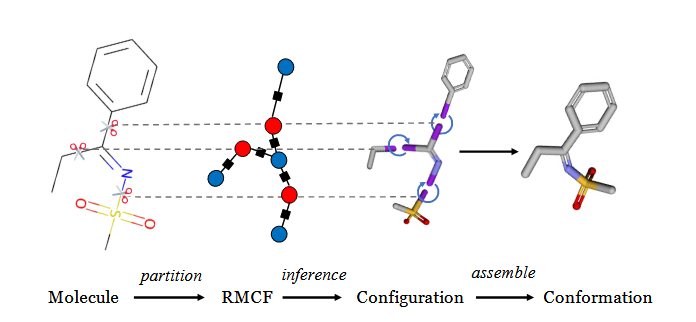
- 构建正则化构象场。依据最少内部自由度(DoF)原则将分子分割。蓝色圆圈代表片段构型,一般来说是低内部柔性的。
- 利用MRF建模。红色圆圈是二面角构型,黑色方块是相邻构型之间的相互作用。利用马尔可夫随机场(MRF)对片段构型和二面角构型的联合概率分布进行建模。
- 推理和采样。推理时无环RMCF采用多年动态规划进行最大后验解码,有环使用LBP算法。采样使用Gibbs采样,每次采样后固定其他节点。采样后用特定距离度量样本差异,通过 K-means 聚类,从每个聚类中随机抽取样本,提升生成构象的多样性。
- 构象组装。将片段和二面角进行组装。
优势
- 通过分子切割减少了构象空间维度,避免生产许多无关变量对模型的影响
- MRF更好地捕捉相邻片段间的关系并对构象不确定性进行建模
数据集
GEOM-QM9和GEOM-Drugs数据集。
- GEOM-QM9
评测方式
- 分子构象的质量和多样性:覆盖分数(COV-R)和匹配分数(MAT-R)
- 预测精度:COV-P和MAT-P。
Zero-Shot 3D Drug Design by Sketching and Generating | NeurIPS 2022
[2209.13865] Zero-Shot 3D Drug Design by Sketching and Generating
文章属于综述里面的分子生成任务下的靶向分子设计的内容。提出零样本3D药物设计方法DESERT(Drug dEsign by SkEtching and geneRaTing)。
目标:
- 主要解决什么问题
- 挑战是什么
- 我们提出的核心方法,与同类问题比较的优势在哪
- 数据集是什么,是否公开
- 评测方式是什么,有无数据集
解决的问题
目前的药物设计中传统方法和深度学习方法都有很多局限性。
挑战是什么
目前的方法都有一些局限性。
- 传统方法遍历大规模药物库,耗时且难以产生新的候选药物
- 现有的深度学习方法依赖稀缺的实验数据,但是蛋白质口袋的生物活性数据大多缺乏,另一些依赖对接模拟,但是这个非常耗时,且准确性不够会影响模型的泛化能力
核心方法
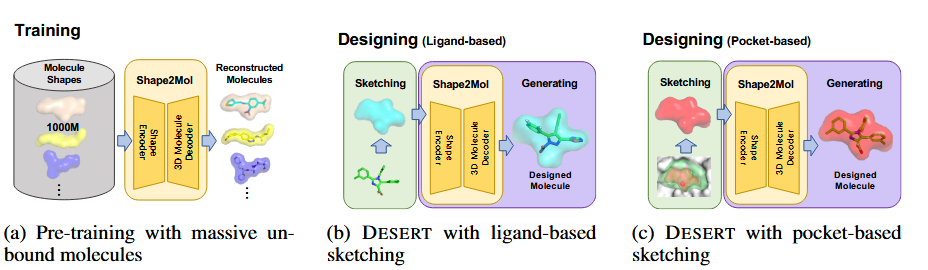
这里提出的方法是DESERT。把药物设计分成草图绘制和生成两个阶段。
- 草图绘制阶段:获取与目标口袋互补的合理的分子形状。
- 有参考配体时,直接使用配体形状
- 在无参考配体时,基于生物学观察从蛋白质口袋中采样合理形状
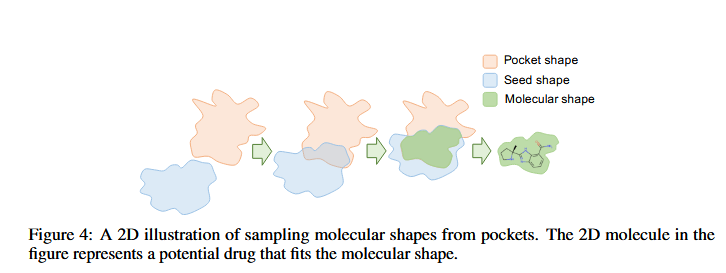
- 生成阶段:通过预训练的SHAPE2MOL模型将形状转换为具体的3D分子。
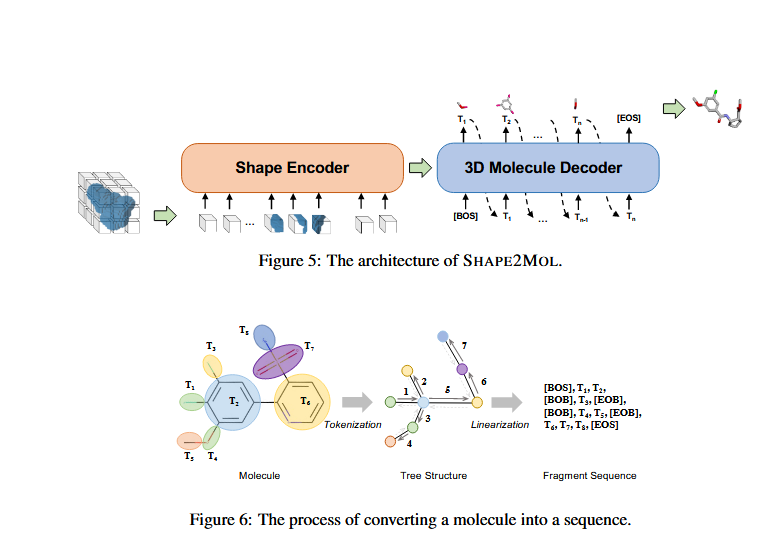
这里SHAPE2MOL将问题形状到分子的生成问题转换为了图像到序列的生成问题。也就是输入3D图像,给出一个序列,表示3D分子。
内部结构是3D拓展后的ViT结构。
优势
- 减少数据和模拟依赖:DESERT 不严重依赖对接模拟,仅在后期可选使用对接进行后处理,同时抛弃了昂贵的实验数据,通过在大规模分子数据库(如 ZINC 数据库)上训练模型,降低了对实验数据的需求,避免了过拟合问题。
- 高效快速:相比基于 MCMC 的 GEKO 模型,DESERT 利用生物知识修剪搜索空间,能更快速地找到较好的解决方案,生成速度比 GEKO 快约 20 倍。
- 生成高质量分子:基于形状的设计方式使 DESERT 能够生成质量更高的分子。
数据集
训练:使用了ZINC数据中的数据对SHAPE2MOL模型进行训练,包含了10亿对分子及其相应形状的数据。
评估模型性能:12 种蛋白质(PDB IDs: 1FKG, 2RD6, 3H7W, 3VRJ, 4CG9, 4OQ3, 4PS7, 5E19, 5MKU, 3FI2, 4J71)相关的数据
评测方式
- 设计结果覆盖的分子空间:唯一性(Uniqueness)、新颖性(Novelty)、多样性(Diversity)、成功率(Success rate)和乘积(Product)
- 高活性分子的能力:通过比较Vina评分的分布,使用 Median Vina Score(Median)来量化分布。
On Pre-trained Language Models for Antibody | ICLR 2023
[2301.12112] On Pre-trained Language Models for Antibody
文章属于综述里面的蛋白质生成里面的抗体生成部分。
目标:
- 主要解决什么问题
- 挑战是什么
- 我们提出的核心方法,与同类问题比较的优势在哪
- 数据集是什么,是否公开
- 评测方式是什么,有无数据集
解决的问题
- 目前的难以探究目前不同的预训练语言模型在抗体任务中的表现。
- 没有引入生物机制在模型之中。
挑战
- 缺乏可靠的抗体特异性基准用于性能评估;
- 对当前蛋白质预训练语言模型(PPLMs)和抗体预训练语言模型(PALMs)的综合研究不足;
- 难以判断引入生物机制是否能真正有益于抗体表示学习;
- 确定预训练表示在实际应用(如药物发现和免疫过程理解)中的作用存在困难
预训练蛋白质语言模型PPLMs:
利用蛋白质序列探索大语言模型。
- 如ProtTrans和ESM-1b将单个蛋白质序列作为输入,使用Transformer架构进行预训练。
- MSA-Transformer/MSA-1b模型通过多序列比对(MSA)作为输入。在结构预测方面,该模型优于 ESM-1b,这表明进化信息有助于蛋白质表征学习。
预训练抗体语言模型PALMs:
- AntiBERTy:提出首个抗体特异性语言模型,对在OAS数据库中的5.58亿条天然抗体序列使用Transformer架构进行预训练。
- Abalang-H/L:恢复抗体序列中缺失的残基上的迁移学习。
- AntiBERTa:在OAS数据库上预训练,并进行微调以用于抗原结合位点位置预测。
核心方法
提出了抗体理解评估(AnTibody Understanding Evaluation,ATUE)基准和包含特定进化信息的EATLM模型。
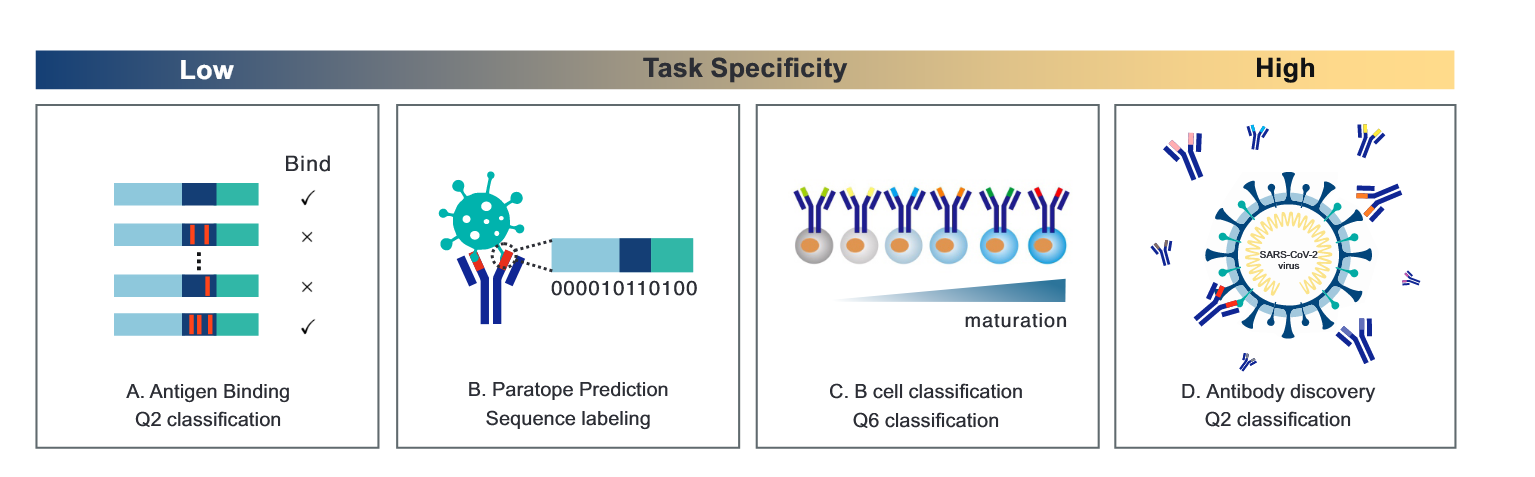
- 创建了一个全面的抗体基准测试工具ATUE。
- 抗原结合预测:二分类序列分类任务,确定抗体的CDR区域能否与特定抗原结合。
- 原因:通过对抗体 CDR 区域的分析,预测其与特定抗原的结合情况,有助于筛选出具有潜在治疗效果的抗体。
- 互补决定区预测:确定抗体序列上的结合位置的序列标注任务为 CDR 片段的每个残基预测 0/1 标签。
- 原因:确定抗体与抗原的结合位置,有助于深入理解抗体与抗原的相互作用机制。
- B 细胞成熟分析:是一个 6 分类任务,区分 B 细胞抗体序列的成熟阶段,每个序列属于 {未成熟、过渡、成熟、浆细胞、记忆 IgD+、记忆 IgD-} 中的一种。
- 原因:有助于理解免疫进化过程中的机制。
- 抗体发现:是一个二分类序列分类任务,区分哪个抗体直接对 SARS-CoV-2 结合负责。
- 原因:从大量抗体中找出能与 SARS-CoV-2 结合的抗体,对于开发针对该病毒的治疗方法意义重大。
- 抗原结合预测:二分类序列分类任务,确定抗体的CDR区域能否与特定抗原结合。
- 得出了关键观察结果,提供了如何更好地表示抗体的一些指导方针。
- PPLMs 在与结构高度相关的抗体任务中表现良好,但在具有高抗体特异性的任务中表现不佳
- 在大多数情况下,PALMs 在预训练数据较少时表现得与 PPLMs 一样好甚至更好
- 通过结合进化过程可以改进 PALMs,但来自 MSA 的进化信息并不总是对抗体任务有益
- 探究引入生物机制对模型的影响,提出EATLM模型。
- 在传统掩码语言建模(MLM)的基础上,引入两个新的预训练目标以模拟抗体进化的生物机制。
- 祖先种系预测AGP
- 突变位置预测MPP
- 在传统掩码语言建模(MLM)的基础上,引入两个新的预训练目标以模拟抗体进化的生物机制。
优势
ATUE基准涵盖了多个具有不同特异性的真实任务,能更加全面地评估模型。
对于EATLM模型来说,引入了AGP和MPP两个预训练目标之后,有以下优势:
- 抗原结合预测:对AUC和F1指标有改进
- 表达预测:在F1和MCC指标上优于其他模型
- B细胞成熟分析任务:显著优于其他PALM模型
- 抗体发现任务:是识别所有钱再结合物最有效的方法,确定了11种潜在的SARS-CoV-2结合抗体,展示了在实际应用中的潜力。
数据集
- 抗原结合预测
- Mason等人(2021)的数据集
- 互补决定区预测
- 使用从 Liberis 等人(2018)收集的包含 1662 个 CDR 片段的数据进行研究。由于只有部分抗体来自进化,所以该任务具有中等特异性。
- B细胞成熟分析
- 数据集来自 Mroczek 等人(2014),有 6 个成熟阶段的 88094 个序列。特异性高,抗体进化与 B 细胞成熟高度耦合。
- 抗体发现
- 研究人员收集了 133 名 SARS-CoV-2 患者和 87 名健康人的抗体序列,按照特定流程处理数据,并与 CoV-AbDab 数据库中的序列匹配,以确定潜在的结合抗体。由于来自同一疾病的抗体具有强烈的趋同种系信号,所以该任务特异性高。
评测方式
对于不同任务,分别进行评估:准确度ACC、马修斯相关系数MCC、F1值、AUC。
Learning Harmonic Molecular Representations on Riemannian Manifold | ICLR 2023
[2303.15520] Learning Harmonic Molecular Representations on Riemannian Manifold
这篇文章所属的领域属于分子生成领域中的表示学习。
解决的问题
- 现在的基于欧几里得空间的分子表示方法需要借助等变网络保证分子在表示旋转和平移的时候的一致性,这里的等距变换群就是E(3)/SE(3)。
- 目前的分子表示学习多采用自下而上,难以提供不同分辨率的特征。
挑战
- 设计一种绕过等变要求,并且能够准确编码3D分子结构的表示。
- 开发在不同分辨率下为不同任务提供合适特征的多分辨率消息传递机制,特别是复杂的大分子。
核心方法
用分子表面的拉普拉斯 - 贝尔特拉米(Laplace - Beltrami)特征函数来表示分子,在 2D 黎曼流形上实现多分辨率的分子几何和化学特征表示,并引入谐波消息传递方法进行高效的谱消息传递。
大模型的解释:
- 改变表示空间:传统方法在 3D 欧几里得空间中编码分子结构,为保证分子表示在旋转和平移时的正确性,需要借助等变网络。而该方法将分子表示在 2D 黎曼流形上。可以把黎曼流形想象成一个可以弯曲、变形,但局部性质类似欧几里得空间的特殊空间。在这个空间上,分子的表示天生就具有旋转和平移不变性。就好比把分子放在一个有弹性但又有自身规律的 “网” 上,无论分子怎么旋转、平移,这个 “网” 对分子的描述都不会改变,不需要额外的等变网络来调整,从而绕过了等变要求。
- 利用拉普拉斯 - 贝尔特拉米特征函数:分子表面可看作黎曼流形,拉普拉斯 - 贝尔特拉米(LB)特征函数是这个流形的固有属性。不同的分子表面有不同的 LB 特征函数,它们就像分子的 “指纹”,能反映分子的形状和结构特点。这些特征函数在刚性变换下保持不变,所以可以用来准确编码分子结构。例如,我们可以把分子表面想象成一个有很多不同纹理的曲面,LB 特征函数就像是描述这些纹理分布规律的工具,不管分子怎么转动、移动,这些纹理的分布规律是不变的,通过分析这些规律就能准确编码分子结构。
- 多分辨率表示与信息传递:通过对 LB 特征函数的线性组合,可以实现分子表面的多分辨率表示。不同频率的 LB 特征函数可以捕捉分子不同尺度的特征,低频部分反映分子的整体、大致的形状,高频部分则能体现分子的细节特征。在进行信息传递(类似消息在分子表面不同区域传播)时,利用基于 LB 特征函数构建的谐波消息传递机制,能在不同尺度上传播信息。这就好像在一个城市中,有不同规模的道路来传递信息,主干道(低频特征)传递整体的、大致的信息,小巷(高频特征)传递详细的、局部的信息,从而全面、准确地编码 3D 分子结构
优势
- 在2D黎曼流形上的分子天然具有旋转和平移不变性,无需依赖等变网络
- 采用自上而下的方式,能提供多分辨率特征
- 分子形状定义了黎曼流形,原子构型决定流形上的相关函数,更全面反映分子性质
数据集
- QM9:小分子性质回归任务
- 配体结合口袋数据集,数据划分方式参考这里
- 刚性蛋白质对接数据集作为训练集,Docking Benchmark 5.5作为测试集。
评测方式
- QM9 小分子性质回归:通过计算预测结果与真实值的平均绝对误差(MAE)来评估模型性能,对比其他不变性和等变网络模型,如 SchNet、NMP 等。
- 配体结合口袋分类:使用平衡准确率评估模型预测蛋白质口袋结合配体类型的能力,与 MaSIF-ligand 模型对比。
- 刚性蛋白质对接:采用 Complex RMSD、Interface RMSD、DockQ 和成功率等指标评估对接性能。Complex RMSD 和 Interface RMSD 衡量预测结构与真实结构的偏差;DockQ 是基于多个标准化标准的综合评分;成功率表示预测结果达到 “可接受” 或更高水平的比例 。
Coarse-to-Fine: a Hierarchical Diffusion Model for Molecule Generation in 3D | ICML 2023
[2305.13266] Coarse-to-Fine: a Hierarchical Diffusion Model for Molecule Generation in 3D
文章属于综述里面的分子生成中的3D分子生成。
解决的问题
现有的3D分子生成方法在生成大尺寸分子的时候存在结构质量差的问题。
挑战
- 自回归模型:按照人工设定的顺序逐个生成原子,如同语言生成过程。但分子在 3D 空间具有自然的几何结构,这种方法引入的人为顺序与分子的自然结构不匹配,并且会产生规模和误差累积问题。
- 非自回归模型:原先的原子级生成方法虽然灵活性较高,但是在片段级生成时,由于化学价态限制,片段冲突常见,且避免片段冲突的复杂度高,随着结构尺寸增加,复杂度呈指数上升,难以获得可靠的分子结构。
核心方法
提出基于分层Diffusion的分子生成方法Hierarchical Diffusion-based 模型(HierDiff)。
将3D分子生成问题看作约束生成问题。
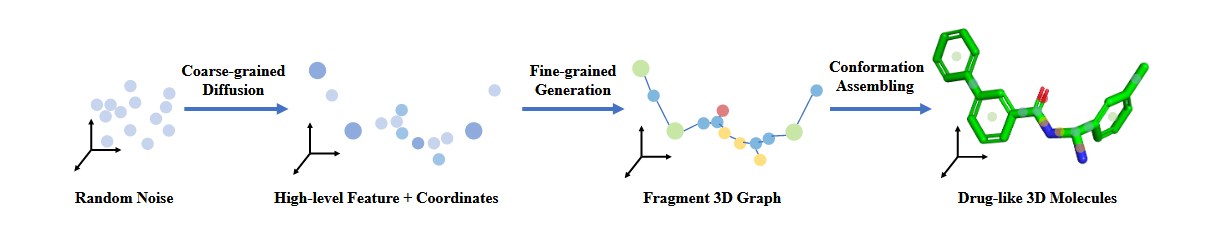
- 先通过等变Diffusion过程生成粗粒度分子几何结构,其中每个粗粒度节点反映分子中的一个片段
- 通过消息传递过程和一个新设计的迭代细化采样模块,将粗粒度节点解码为细粒度片段
- 将细粒度片段组装起来,得到完整的原子分子结构
详细解释
- 粗粒度片段扩散:HierDiff 将 3D 分子生成视为约束生成问题,首先定义粗粒度节点的表示,包括不变化学特征和等变位置特征。通过精心设计化学特征(如基于属性和元素的特征)和位置特征(使用中心坐标),利用扩散模型生成粗粒度片段表示及其笛卡尔坐标。在这个过程中,通过特殊设计的初始分布和转移核,保证模型的 SE (3) 不变性,从而有效生成合理的粗粒度分子几何结构。
- 细粒度片段生成:基于生成的粗粒度节点,通过一系列步骤生成细粒度片段类型和边。具体包括选择焦点节点、预测新边、确定细粒度片段类型以及迭代细化。利用消息传递神经网络和迭代细化模块,不断纠正细粒度节点中的偏差,提高生成分子的真实性。
- 组装成原子构象:根据细粒度生成过程确定的节点和链接关系,选择合适的原子合并方式构建原子级构象。利用 RDkit 生成局部构象,并通过 Kabsch 算法计算旋转矩阵和平移向量,将局部构象对齐到采样的中心位置,逐步生成完整的原子构象。
优势
与同类方法相比,HierDiff 能保持非自回归方法的全局建模特性,显著降低寻找可连接片段的复杂度,有效避免片段冲突,生成的分子更具现实性和药物样属性,在多个评估指标上优于现有方法。
数据集
使用GEOM-Drugs和CrossDocked2020数据集。
评测方式
- 药物相似性
- QED
- 逆向合成可及性RA
- 药物化学过滤器MCF
- 合成可及性分数SAS
- LogP
- $\Delta$LogP
- 分子重量MW
- 构象质量
- 计算覆盖度Cov
- 匹配度Mat
Accelerating Antimicrobial Peptide Discovery with Latent Structure | SIGKDD 2023
[2212.09450] Accelerating Antimicrobial Peptide Discovery with Latent Structure
这篇文章属于综述下面的蛋白质生成下的肽设计部分。
解决的问题
加速抗菌肽(AMP)的发现。现有的深度学习模型只考虑序列属性,但是忽略了结构对于活性的关系。
挑战
目前深度学习模型只考虑序列特征,忽略结构-活性关系。结构对于肽的活性具有重要影响,但是却没有应用。
核心方法
提出 Latent Sequence-Structure 模型(LSSAMP)。
- 将序列特征和二级结构映射到潜空间中
- 采用VQ-VAE为每个位置分配一个潜在变量
- 通过在潜空间采样生成理想序列组成和结构的肽
优势
考虑了序列和结构信息,肽的抗菌性更强
数据集
- Universal Protein Resource(UniProt)中的蛋白质序列和通过ProSPr预测的二级结构
- Antimicrobial Peptide Database中的抗菌肽数据集
评测方式
- 自动评估指标
- 使用开源的AMP预测工具估计生成序列的AMP概率。
- 依据电荷、疏水性、疏水矩这三个对AMP机制至关重要的序列属性评估生成性能。
- 通过唯一性、多样性和相似性来衡量生成的肽的新颖性。
- 湿实验室实验
- 通过上面的自动评估指标从5000个肽中筛选出21个肽进行合成实验
- 合成了之后在培养皿中采用肉汤微量稀释法测定最小抑菌浓度(MIC),从而验证。
Equivariant Flow Matching with Hybrid Probability Transport for 3D Molecule Generation | NeurIPS 2023
这篇文章属于分子生成领域中的3D分子生成。
解决的问题
3D 分子生成需要同时确定原子类型和坐标。
目前的模型,特别是Diffusion方法存在采样速度低和概率动力学不稳定的问题。
挑战
现在的扩散模型存在概率动力学不稳定和采样速度低的问题。
核心方法
提出等变流匹配(EquiFM)框架。
- 引入等变最优传输(Equivariant Optimal-Transport)引导原子坐标的生成概率路径。这方法蕴含最小化坐标变化的先验,能稳定训练并提升生成性能。
- 基于信息量构建混合生成路径解决模态不一致问题:根据不同组件的信息量差异,设计不同的生成概率路径,形成混合生成路径。
- 原子特征空间包含多种数据模态,如电荷、原子类型和坐标分别属于离散、整数和连续变量。
- 利用 ODE 参数化模型提升推理效率:模型基于连续归一化流,由 ODE 参数化。相比Diffusion使用的SDE提高了推理效率。
优势
- 稳定
- 效率高
- 生成分子质量更好
数据集
QM9、GEOM-DRUG数据集
评测方式
- 分子建模与生成
- 通过预测键类型评估生成分子的化学可行性,计算原子稳定性、分子稳定性、有效性、唯一性等指标衡量生成质量
- 条件分子生成
- 在 QM9 数据集上测试模型根据给定属性生成分子的能力,通过计算生成分子属性值与目标属性值的平均绝对误差(MAE)来评估性能。
Unified Generative Modeling of 3D Molecules with Bayesian Flow Networks | ICLR 2024
这篇文章属于分子生成领域的3D分子生成部分。
解决的问题
生成模型在应用于3D分子几何生成时面临的挑战,尤其是多模态和噪声敏感性问题。
挑战
- 多模态
- 分子几何的原子级描述依赖多种数据形式,不同模态数据的统一处理较为困难。
- 噪声敏感性
- 微小的坐标噪声就可能导致分子层面的信号急剧下降,影响模型性能
核心方法
提出GeoBFN。
贝叶斯流网络BFNs
- 假设:数据样本的信息应沿着潜变量的马尔科夫链逐步增加,且信息变化尽可能平滑。
- 基于引入噪声变量的潜变量模型,通过优化变分下界学习概率分布,在参数空间操作以保证信息变化平滑
- 统一概率建模
- 使用统一的概率建模公式处理分子几何中的不同模态。
- 将3D分子表示为$g=<x,h>$,其中$x$为原子坐标矩阵,$h$包含原子类型和原子电荷等节点信息。
- 通过EGNN对输出分布进行参数化
- 保持SE(3)不变性
- 零质心空间约束下,通过设计满足特定条件的概率模型,使似然函数 \(p_{\phi}\) 具有平移和旋转不变性,
- 克服噪声敏感性
- GeoBFN 在参数空间中通过贝叶斯更新过程来降低方差。在更新中,噪声程度较低的样本会被赋予更小的权重。
- 优化离散变量采样
- 使用
NEAREST_CENTER函数将输入和中心桶进行比较,并为每个输入值返回值返回最近的中心。
- 使用
优势
- 生成质量上表现卓越。
- 可以在任意采样步数下达到效率和质量的最优平衡
- 采样效率较高
数据集
QM9和GEOM-Drugs
评测方式
- 无条件分子生成
- 条件分子生成
Multimodal Molecular Pretraining via Modality Blending | ICLR 2024
这篇文章属于分子生成领域中的表示学习方向。
解决的问题
现在的多模态分子预训练方法在对齐2D和3D模态时,仅进行粗粒度分子级对齐,未充分挖掘内在关系。
挑战
- 准确捕捉2D和3D分子中原子关系的内在联系
- 充分整合多模态信息的模型架构和训练方法
核心方法
提出blend-then-predict自监督学习方法。
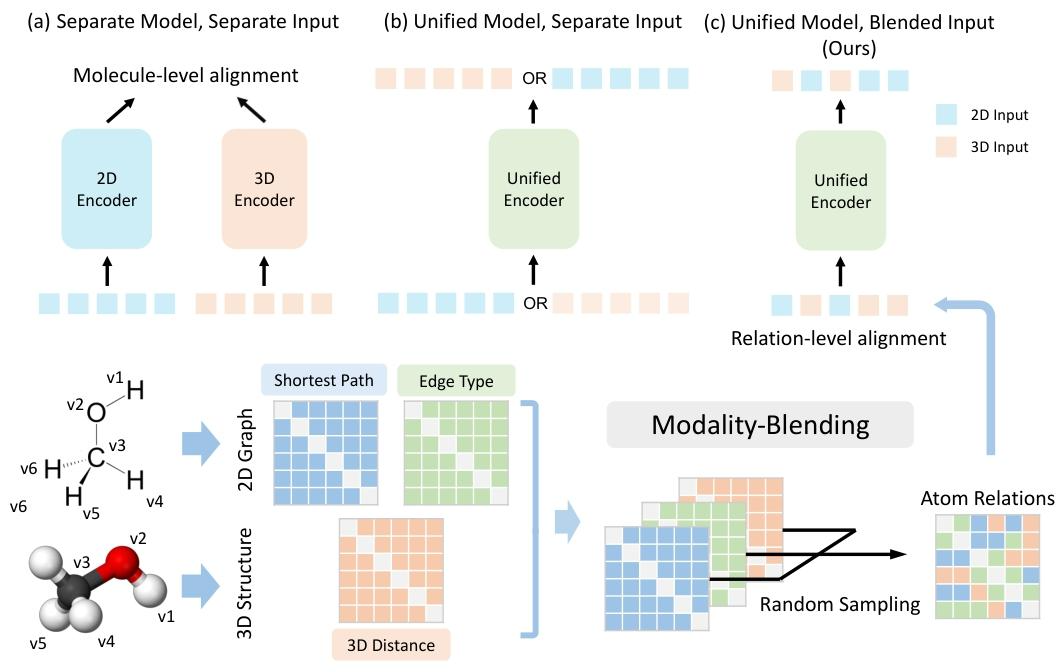
- 将不同模态的原子关系混合成统一矩阵进行联合编码,注入到Transformer的自注意力模块中
- 再恢复特定模态信息
优势
能在细粒度原子级别对齐和整合2D和3D模态,全面描绘分子
数据集
预训练使用 PCQM4Mv2 数据集,来自 OGB Large - Scale Challenge.
评估在多个公开数据集上进行
- MoleculeNet(用于 2D 分子性质预测,涵盖多种分子性质相关数据集)
- QM9 量子性质数据集(包含 13.4 万个小有机分子,用于 3D 任务评估)
评测方式
- 2D任务:使用支架分割(scaffold split)方式划分数据集
- 分类任务以ROC-AUC分数为指标
- 回归任务以RMSE为指标
- 3D任务:随机划分验证集和测试集
- 以MAE为评估指标
Learning Multi-view Molecular Representations with Structured and Unstructured Knowledge | SIGKDD 2024
这篇文章属于分子生成领域的表示学习方向。
解决的问题
现有分子表示学习(MRL)模型在学习多视图分子表示时存在不足。难以从化学结构、生物医学文本和知识图谱等异构源中有效捕捉分子知识,且无法充分利用不同视图间的共识和互补信息,不能很好地适应不同应用场景。
多视图分子表示指的是从不同角度捕捉分子信息,如从微观角度,分子有原子、化学键等;从宏观角度,分子的晶体结构、物理状态等。
挑战
- 现有分子表示学习(MRL) 模型需将视图信息显式融入表示中,以适应广泛应用,但此前模型多通过 “包装文本” 或微调隐式整合,影响对不同视图知识关系的理解
- 要处理分子结构、生物医学文本和知识图谱等质量和数量各异的信息源的异质性,以往将知识图谱转换为文本的方法可能因预训练数据分布不均衡引入偏差。
核心方法
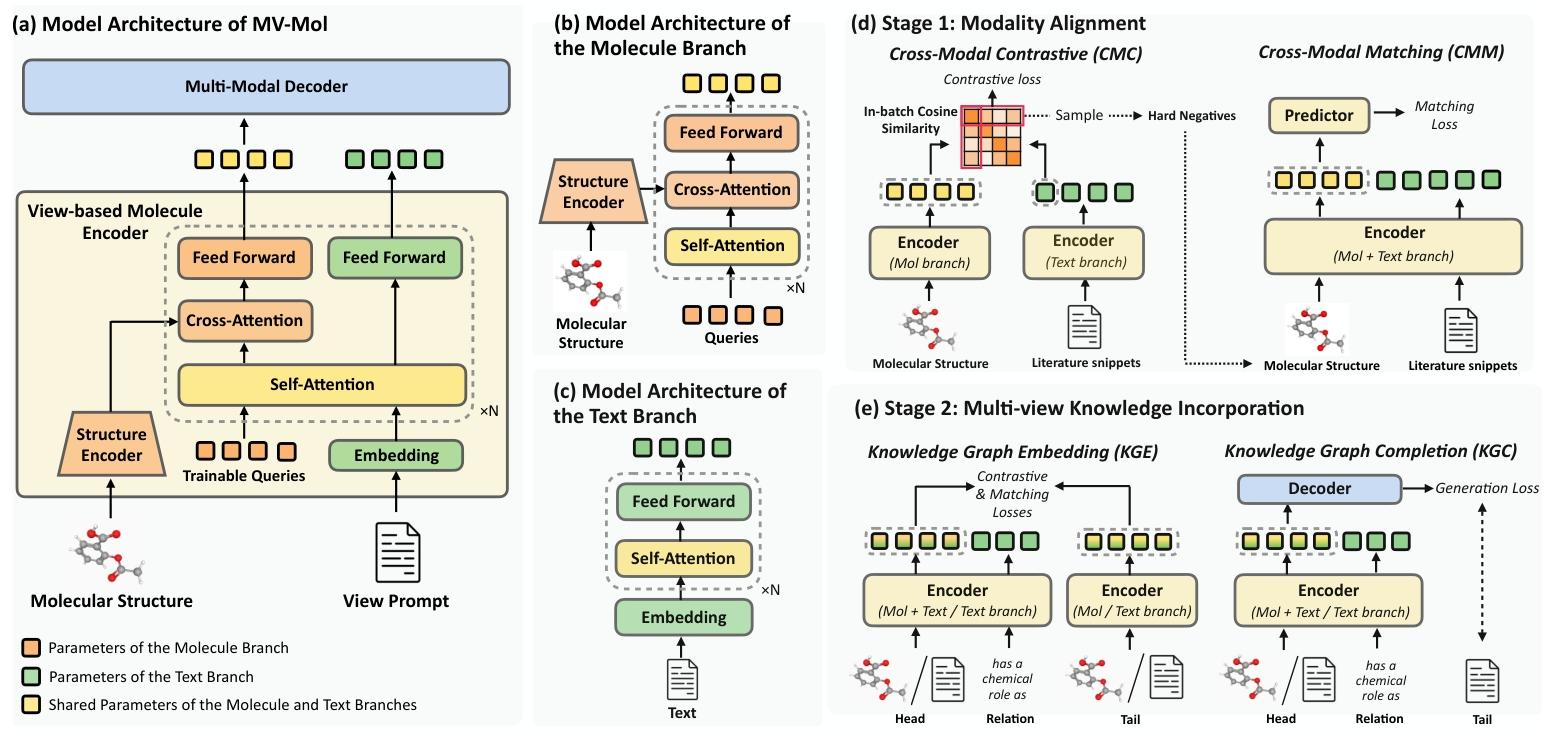
提出 MV-Mol 模型
- MV-Mol 模型架构
- (a) 基于视图的分子编码器:此编码器是获取视图相关分子信息的关键组件,输入为分子结构和文本提示。
- (b)分子分支
- 结构由\(M=(V, E, C)\)表示,利用预训练的 Uni-Mol 进行编码,将其转化为原子的特征表示\(z(a)\)。
- Q-Former 的分子分支以 K 个可训练的查询向量作为输入嵌入,借助跨注意力机制,在每隔一个 Transformer 层时,从原子表示\(z(a)\)中提取关键信息。
- (c)文本分支
- 提示\(T=[x_{1}, x_{2}, \cdots, x_{L}]\)代表不同的分子视图。
- Q-Former 的文本分支对文本提示T进行处理。
- Q-Former 两个分支的自注意力层共享,使分子表示能够融合不同视图的信息,最终输出基于视图的分子表示。
- (d)模态对齐
- 通过跨模态对比和跨模态匹配进行模态对齐。
- 跨模态对比损失用于最大化分子结构与文本表示间的互信息,
- 跨模态匹配损失则通过预测分子结构和文本是否对应同一分子,来培养模型对两者的细粒度理解
- (e)多视图知识融合
- 将关系建模为一种文本提示,从特定视图约束分子知识。
- 设计知识图谱嵌入和知识图谱不全目标来实现多视图知识融合。
- (b)分子分支
- 多模态解码器:采用 BioT5 的解码器作为多模态解码器。
- 输入是基于视图的分子编码器输出。
- 通过因果生成,将输入转化为自然语言文本,实现对分子表示的自然语言解释 。
- (a) 基于视图的分子编码器:此编码器是获取视图相关分子信息的关键组件,输入为分子结构和文本提示。
优势
MV-Mol 能更好地捕捉不同视图的分子知识。
- 在分子属性预测任务上,比最先进的 Uni-Mol 模型平均绝对增益 1.24%
- 在跨模态检索任务中,相比最佳基线模型,top-1 检索准确率平均提高 12.9%
- 在跨模态生成任务中预测也更准确。
数据集
预训练采用大规模分子-文本对和知识图谱。
- 分子 - 文本对通过对 350 万篇科学出版物进行命名实体识别和实体链接获得
- 知识图谱由多个公共数据库合并构建。
下游实验使用多个数据集
- 用于分子属性预测的 MoleculeNet 中的 8 个分类数据集
- 用于跨模态检索的 PCdes 和 MVST 数据集
- 用于跨模态生成的 ChEBI-20 数据集等。
代码和数据可在https://github.com/PharMolix/OpenBioMed获取。
评测方式
- 在分子属性预测中
- 采用 Scaffold split 划分数据集
- 在多个数据集上微调模型并报告 AUROC 分数
- 跨模态检索包含结构到文本和文本到结构检索两个子任务,
- 使用 PCdes 和 MVST 数据集
- 按 Scaffold split 划分
- 报告 MRR(平均倒数排名)和 Recall at 1/5/10
- 跨模态生成包括结构到文本生成和文本到结构生成
- 在 ChEBI-20 数据集上按原始划分进行实验
- 采用 BLEU、ROUGE、METEOR 等指标评估分子字幕任务
- 用精确率、有效率等指标评估文本到分子生成任务
MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space | ICML 2024
这篇文章属于分子生成领域的靶向药物分子设计方向中的基于结构的药物设计(SBDD)。
解决的问题
靶向分子设计有两种,一种是基于配体的药物设计(LBDD),另一种是基于结构的药物设计(SBDD)。LBDD利用目标蛋白质的氨基酸序列,借助已知的配体特征来构建;SBDD利用目标蛋白质的三维结构来设计。
当前基于SBDD生成模型在生成分子时,常出现不符合要求的情况。
生成的分子不能同时满足高亲和力、良好的类药性质和合理的 3D 构象这几个关键标准,产生假阳性结果,阻碍了 SBDD 模型在实际中的应用 。
挑战
- 分子模式坍塌:自回归模型在生成分子时倾向于产生有限数量的特定(子)结构,从化学和几何角度来看,其生成的独特分子比例较低,对某些环结构存在偏好,且在模拟不同键类型的键长时表现不佳,无法有效捕捉参考分布的多模态特征。
- 混合连续 - 离散空间:Diffusion模型虽然通过非自回归生成在一定程度上缓解了模式坍塌问题,但混合连续 - 离散空间使得模型难以准确捕获分子的复杂数据流形。在这个空间中进行去噪时,不同模态之间的不一致性会导致生成的分子存在高应变和不可行的情况,中间噪声潜在值容易超出流形范围。
核心方法
提出MolCRAFT模型,是首个在连续空间运行的SBDD模型。
- 特点
- 统一的 SE-(3) 等变生成模型
- 在连续参数空间中进行分子生成
- 实现
- 统一参数化:将连续原子坐标和离散原子类型分别进行参数化。
- 不同模态噪声处理:由于参数的连续性,即使对于离散原子类型也能应用连续噪声。
- SE(3)等变网络:使用 SE-(3) 等变网络对蛋白质 - 分子复合物的相互作用进行建模,确保模型在平移和旋转下的不变性。
- 噪声减少采样策略:
- 传统的采样方式在每个时间步都对连续原子坐标和离散原子类型进行采样,易引入过多噪声。
- MolCRAFT 设计了在参数空间内的噪声减少采样策略,用估计的\(\hat{m}=[\hat{x},\hat{v}]\)(\(\hat{v}\)直接采用连续输出的类别值而不采样)直接更新下一步的参数。
优势
- 结合亲和力。
- 能达到参考水平的 Vina 评分(-6.59 kcal/mol),远超其他强基线模型
- 构象稳定性
- 在模拟局部模式时表现出色,在键长和角度分布上排名第一,且生成的配体 - 蛋白质复合物中的冲突更少,重新对接后的 RMSD 表现最佳,46% 的生成分子在无需力场优化或重新对接的情况下就接近准确的对接姿势
- 采样效率更高
- 速度更快
数据集
使用 CrossDocked 数据集,经过基于 RMSD 的过滤和 30% 序列同一性拆分后,得到 100,000 个训练对和 100 个测试蛋白质。
评测方式
- 结合亲和力
- 构象稳定性
- 类药性质
- QED
- 合成可及性SA
- 多样性Div
- 整体评估
- 结合可行性(合理亲和力+构象稳定的分子比例)
- 成功率(满足Vina Dock、QED和SA阈值)
- 生成效率
ESM All-Atom: Multi-scale Protein Language Model for Unified Molecular Modeling | ICML 2024
这篇文章属于蛋白质生成领域中的表示学习领域。
解决的问题
当前蛋白质语言模型主要在残基尺度运行,无法提供原子尺度信息。
挑战
- 统一分子建模难题:残基和原子尺度使用的词汇表不兼容,直接在原子尺度对蛋白质进行表示和预训练效率低下,难以实现有效的统一分子建模。
- 位置编码设计困难:设计合适的位置编码来准确描述同一蛋白质中残基和原子之间的关系颇具挑战,涉及残基与残基、残基与原子、原子与原子之间的多种关系,而现有蛋白质语言模型的编码方法无法满足需求。
核心方法
提出ESMAA(ESM All-Atom),实现原子尺度和残基尺度统一分子建模的新方法。
- 在多尺度代码转换蛋白质序列上进行预训练
- 利用多尺度位置编码来捕捉残基和原子之间的关系
优势
ESM-AA 能同时处理残基和原子尺度信息
- 在蛋白质 - 分子任务上表现更优。
- 如在酶 - 底物亲和力回归、药物 - 靶点亲和力回归等任务中超越了其他模型,实现了最先进的结果。
- 在蛋白质任务和分子基准测试中也有良好表现
数据集
预训练数据集
- 蛋白质
- AlphaFold DB 数据集: 800 万个由 AlphaFold2 预测的高置信度(PLDDT > 90)蛋白质序列和结构
- 小分子
- 使用 Zhou 等人提供的数据集:含有 1900 万个分子和 2.09 亿个由 ETKGD 和默克分子力场生成的构象
评测方式
- 蛋白质 - 分子任务:在酶 - 底物亲和力回归、药物 - 靶点亲和力回归和酶 - 底物对分类任务上进行微调评估,将模型预测结果与实验数据对比,使用均方误差(MSE)、决定系数(R²)、皮尔逊相关系数、准确率(ACC)、马修斯相关系数(MCC)、受试者工作特征曲线下面积(ROC-AUC)等指标衡量性能。
- 蛋白质任务:通过二级结构预测和无监督接触预测任务测试模型对蛋白质结构的理解能力,使用准确率等指标评估,且模型输入为纯残基序列。
- 分子任务:利用标准分子基准测试 MoleculeNet 中的任务,如分子性质分类和回归任务,使用平均绝对误差(MAE)、AUC 等指标评估模型性能。
Mol-AE: Auto-Encoder Based Molecular Representation Learning With 3D Cloze Test Objective | ICML 2024
这篇文章属于分子生成领域中的3D分子表示学习方向。
解决的问题
- encoder-only model在预训练和下游任务目标之间存在不一致性,导致预训练学到的特征在下游任务中迁移性差
- 坐标去噪目标会引发训练不稳定以及引入不真实噪声,同时原子坐标在该过程中作为内容和标识符的双重角色产生冲突,影响模型性能。
挑战
- 克服预训练和下游任务目标不一致带来的难题,使模型在不同阶段学到的特征能有效应用于实际任务
- 解决坐标去噪导致的训练不稳定和原子标识符混乱问题,让模型能够稳定训练并准确学习分子结构信息。
核心方法
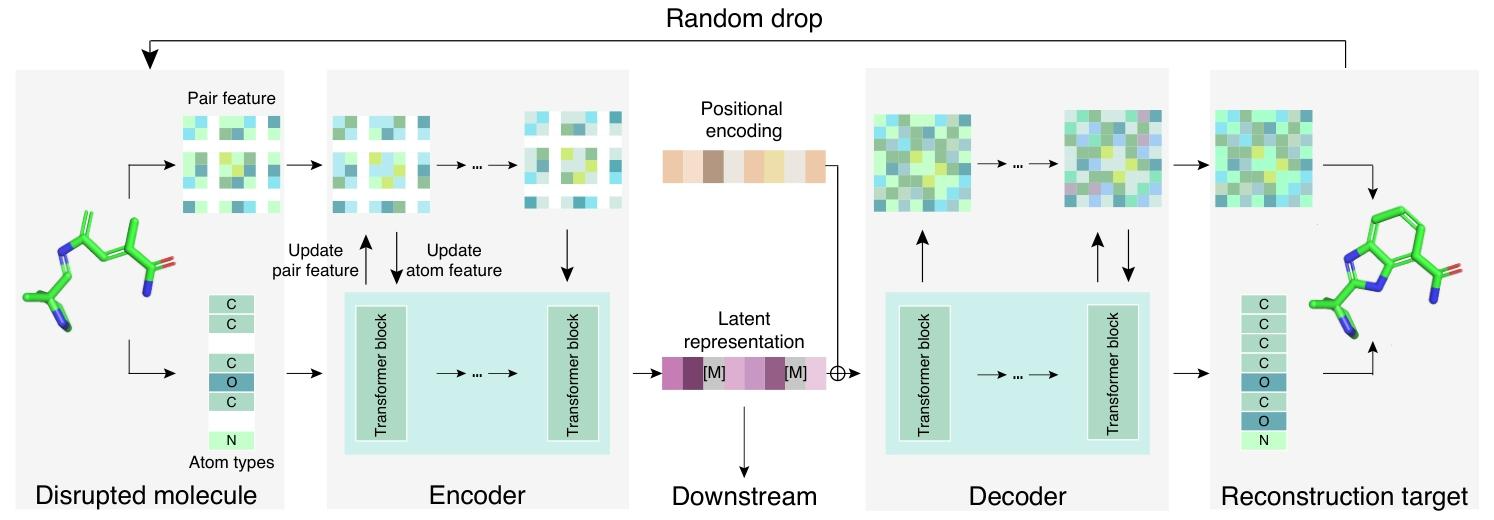
提出 MOL-AE 模型,核心包含基于 Transformer 的 3D 信息感知自编码器结构以及 3D Cloze Test 目标。
- 整体架构:
- MOL-AE 模型处理 3D 分子时,主要聚焦 3D 结构和原子类型信息。
- 原子类型建模:可借助原子 MLM 目标轻松实现
- 3D 结构的建模:模型由编码器\(q_{\phi}\)、解码器\(p_{\theta}\)构成,且二者均以 Transformer 架构为基础。
- MOL-AE 模型处理 3D 分子时,主要聚焦 3D 结构和原子类型信息。
- 3D 信息感知自编码器
- Transformer Block:Transformer 由多个 Transformer 块组成,每个块包含多头自注意力层和前馈层。此过程能有效捕捉分子信息。
- 3D 感知成对特征:因普通 Transformer 难以处理 3D 信息,MOL-AE 采用将原子对之间的欧几里得距离编码为额外成对特征的方法。帮助模型更好地理解分子的3D结构。
- 编码器和解码器:编码器\(q_{\phi}\)由\(L^{enc}\)层 Transformer 块构成,3D 坐标信息C经其处理后编码为\(X^{L^{enc}}\) ,并将其作为潜在表示Z 。解码器\(p_{\theta}\)由\(L^{dec}\)层 Transformer 块组成,由于 3D 结构已编码在Z中,其输入成对特征初始化为零。
- 3D Cloze Test 目标
- 添加位置编码(PE)到解码器:为解决坐标去噪中原子标识符混乱问题,MOL-AE 在解码器中添加 PE。
- 在打乱坐标时,PE 作为稳定的标识符,帮助模型区分不同原子。同时,仅在解码器添加 PE 可避免引入的顺序信息对编码器学习分子高质量表示产生影响。
- 这里可以类比GPT会添加位置编码。
- 原子丢弃:传统去噪目标可能使模型学习不可靠的噪声分布,MOL-AE 通过随机丢弃部分原子及其坐标(如从输入坐标C中随机移除k行得到\(D(C)\) )来干扰数据,使模型专注于剩余无噪声的子结构,从而更好地学习原子空间关系。
- 添加位置编码(PE)到解码器:为解决坐标去噪中原子标识符混乱问题,MOL-AE 在解码器中添加 PE。
优势
- 缓解了预训练和下游任务目标不一致的问题,提升了特征的迁移能力
- 解决了坐标去噪带来的不稳定训练和原子标识符混乱问题,使模型训练更稳定
- 在多个分子理解任务中性能显著优于当前最先进的 3D 分子建模方法,在分子分类和回归任务的基准测试中取得了优异成绩
数据集
- 预训练数据集:使用 Zhou 等人提供的大规模分子数据集,包含 1900 万个分子和 2.09 亿个构象,由 ETKGD 和 Merck 分子力场生成,每个分子有 11 个随机生成的构象,为提高计算效率,预训练时去除了氢原子,未明确该数据集是否公开。
- 微调数据集:采用广泛使用的 MoleculeNet 基准数据集,包括 9 个分类数据集和 6 个回归数据集,该数据集公开。
评测方式
- 分子分类任务:使用 ROC-AUC 作为评估指标,在 9 个分类数据集上进行实验,数据集为 MoleculeNet 中的相关分类数据集。
- 分子回归任务:采用平均绝对误差(MAE)和均方根误差(RMSE)作为评估指标,在 6 个回归数据集上进行实验,数据集为 MoleculeNet 中的相关回归数据集。通过与多个监督和预训练方法的对比,评估 MOL-AE 的性能。